
Revolutionizing Material Modeling: MIT’s Breakthrough in Predicting Metal Alloy Behavior
In the swiftly progressing fields of aerospace, energy, and computing, the unending pursuit for new materials that assure optimal performance is a constant. But, realistically, the leap from idea to practical use is often hindered by the intricate challenges associated with predicting a material’s behavior. Traditionally, to comprehend a material’s performance, it first needs to be built and tested, which results in substantial cost and time being added to the innovation cycle. Even the highly progressive simulation methods struggle to model the elaborate chemical arrangements present in today’s solid materials.
Breaking Ground with Material Modeling
Led by Rodrigo Freitas, MIT’s TDK Career Development Professor in Materials Science and Engineering, a trailblazing team of researchers has recently pioneered an approach to accurately model metallic substances, regardless of their puzzling chemical configurations. This innovative procedure employs machine-learning models to boost the precision and quickness of material simulations. The researchers made training datasets that cover the vast scope of atomic environments found in chemically unorganized materials, moving beyond the confines of what was thought to be feasible.
Machine Learning: The Future of Material Science
Every material has its own distinctive properties which are determined the unique layout of atomic elements within it. Even materials carrying the same chemical components can display significantly different attributes, influenced by their atomic arrangements. Simulations at an atomic level are necessary to capture these finer details. Machine learning surfaces as a potent tool in constructing these models, especially while working with ordered chemical arrangements. However, the true test comes when modeling chemically disordered phases, a commonly occurring phenomenon in most solid substances.
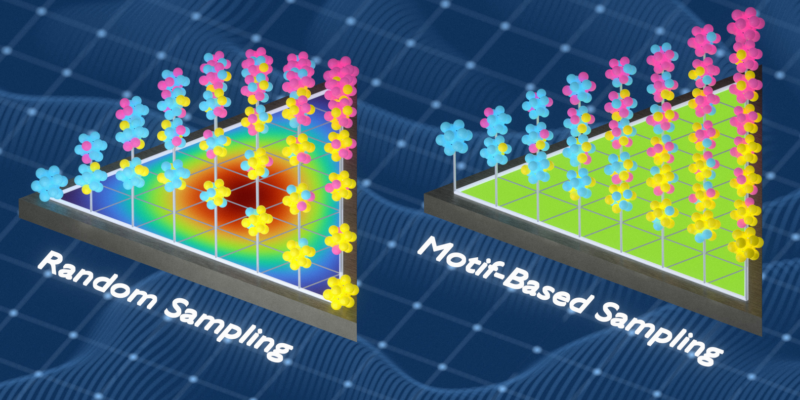
Facing these challenges head-on, Freitas’ team successfully tackled the hurdles posed by chemically unorganized materials, which exhibit a broad array of local chemical environments. They innovatively utilized information theory to create training datasets that more accurately mirror the local chemical environments in such materials.
From Findings to Functional Usage
When their approach was applied to develop machine-learning training datasets for several variant metal alloys, it paid off. Their models managed to outpace larger-scale models from corporate powerhouses like Google and Microsoft. The success was credited to their method’s knack to sense delicate energetic tendencies towards particular local chemical configurations. These are crucial in determining material phases and attributes. Notably, the research was supported by the U.S. Air Force Office of Scientific Research.
But this breakthrough is not limited to scholarly circles. Practical implications abound as well. With the ability to accurately predict phase diagrams – tables that map out stable phases across varying temperatures and chemical compositions – this model has the potential to remarkably impact real-time processing decisions in various industries. The researchers aim to weave these predictions into standard materials design workflows to encourage industry integration that aligns with existing operation protocols.
As industries strive to innovate and optimize, the assimilation of this advanced modeling technology could revolutionize material development and use. Interested readers can explore the original news article from MIT here.
Intrigued by AI automation solutions for your organization? Experience the difference AI can make with implementi.ai.






